科研人員建立原噬菌體de novo預測新算法
中國科學院南海海洋研究所研究員王曉雪團隊建立了一種不依賴于噬菌體基因序列相似性的原噬菌體de novo預測新算法,集成分析流程的工具名為Prophage Tracer。相關研究9月22日在線發表于《核酸研究》。湯開浩博士為論文第一作者,王曉雪為通訊作者。來自天津生物芯片技術有限責任公司的孫亞民博士也參與基因組分析工作。
烈性噬菌體侵染細菌宿主后,大量繁殖,裂解宿主細胞釋放子代噬菌體粒子。溫和噬菌體則是進入宿主細胞內,將自身的DNA整合在宿主的基因組中,隨著宿主細胞的復制而復制。這個過程稱為噬菌體的“溶原化”,整合在宿主基因組中的噬菌體DNA稱為“原噬菌體” (prophage)。特定條件能重新激活原噬菌體使其裂解宿主。目前研究發現在人、小鼠腸道以及珊瑚微生物組中溶原化的溫和噬菌體比裂解狀態更加普遍。溫和噬菌體的溶原-裂解轉換是微生物生態領域的重要科學問題之一。
溫和噬菌體能和宿主形成長期共生關系,其整合切離等可以破壞或恢復宿主的基因正常功能,是宿主基因表達調控的一種重要途徑。研究團隊的前期研究發現希瓦氏菌的原噬菌體CP4So可以在低溫誘導條件下發生切離,是細菌一種重要的適冷機制(1),并且 CP4So的切離受到宿主溫度依賴性的H-NS磷酸化調控(2)。因此精確鑒定細菌中的原噬菌體及其插入位點對于研究溫和噬菌體與宿主的共生關系至關重要。目前鑒定原噬菌體的方法主要依賴利用已知的噬菌體進行序列相似性檢索。但是由于噬菌體的基因組變異快,基于序列相似性檢索方法很難發現未知類型噬菌體。此外,對噬菌體插入位點的預測也不準確,很難區分原噬菌體攜帶的”cargo基因”和宿主基因的邊界。
為了解決上述問題,湯開浩等建立了一種從頭預測原噬菌體的方法(圖1)。該方法主要利用原噬菌體整合切離過程中會產生基因組結構變異基因組序列。這些序列隱藏在細菌基因組、轉錄組測序數據的reads中。通過建立重疊的序列比對方法,追蹤和挖掘埋藏于測序原始數據中reads。只需甄別到1-2條reads就能精確定位原噬菌體(精確到單個堿基)。由于該方法不依賴于序列相似性,因此能夠預測到未知的噬菌體。通過挖掘,成功的在珊瑚共附生細菌中鑒定到九個溫和噬菌體,其中兩個為新穎的溫和噬菌體。
本研究工作得到國家杰出青年科學基金、基金委水圈微生物重大研究計劃重點項目、面上項目、國家重點研發計劃等項目的共同資助。
相關論文鏈接:https://academic.oup.com/nar/advance-article/doi/10.1093/nar/gkab824/6374144
Prophage Tracer下載地址:https://github.com/WangLab-SCSIO/Prophage_Tracer
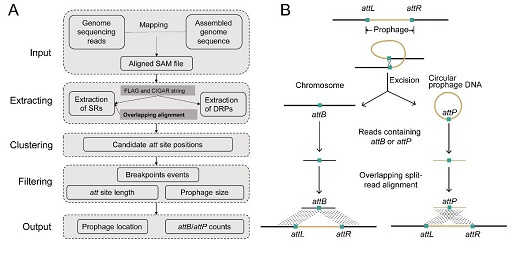
圖1 Prophage Tracer流程


